Spial help page
Spial overview
Spial (specificity in alignments) is a web-server that takes a pair of multiple sequence alignments as input and produces an output that highlights positions which are conserved in both alignments (the consensus), and positions which are specific to either alignment. Given a structure, Spial maps the highlighted positions to color-coded residues on the structure.
About Spial
Spial was developed by Arthur Wuster, A.J.Venkatakrishnan, Gebhard Schertler and M. Madan Babu at MRC Laboratory of Molecular Biology. It has been tested by numerous structural biologists based at this institute.
The following tools and services have inspired Spial or offer similar functionalities:
- Two Sample Logo, developed by Vladimir Vacic, creates logos that are similar to the logos that Spial produces.
- ConSurf is a server that colours the residues of protein structures according to how conserved they are across an alignment of homologous sequences. It was developed in the group of Nir Ben-Tal.
- WebLogo, developed in the group of Steven Brenner, is a tool to visualise multiple sequence alignments as logos. Spial logos are made using WebLogo code.
Spial input
Spial accepts two types of inputs: sequences and alignments.
The sequences input is aligned using MUSCLE and split to produce two alignments (hereon referred to as alignments A and B). Alternatively, Spial directly accepts two sets of alignments as input. The two input alignments have to be of the same length and the positions in both alignments have to correspond.
Spial algorithm
For each amino acid at each position in the two input alignments A and B, Spial decides whether the amino acid is consensus or not.
In order for an amino acid to be consensus, it has to be present above a certain proportion in both alignments.
This proportion can be specified by using the consensus threshold option.
In the example below, the consensus threshold was 0.35. Positions 1, 2, 7, 9, and 10 had amino acids that were present above this proportion in both alignments.
# pos 1234567890
seq1.1 AD-RVAT-SH
seq1.2 ADYKV-S-SH
seq1.3 ADY-VVS-SS
seq1.4 AEYGVIS-SS
seq1.5 VEYHVMT-SS
seq2.1 AEWTLMTPSM
seq2.2 AEWTILTPSM
seq2.3 AEWTMITPPS
seq2.4 AEWTGFTPPS
4631206357
CCSSS0CSCC
Next, Spial decides whether there are amino acids that are specific for any of the two alignments, but not for the consensus.
For this, a non-consensus amino acid has to be present above a certain proportion in either alignment.
This proportion can be specified by using the specificity threshold option.
In the example above, the specificity threshold was 0.35.
Positions 2, 3, 7, 8, and 10 had non-consensus amino acids that were present above this proportion in alignment A.
Positions 3, 4, 8, 9, and 10 had non-consensus amino acids that were present above this proportion in alignment B.
For more on what the numbers and letters in the two rows below the alignment mean, see position types.
Examples
We have pre-computed two examples of Spial output.
The first example has STAT5a as alignment A and STAT4 as alignment B. These are two families of the signal transducer and activator of transcription proteins. Unphosphorylated STAT5a dimerises in a way that is different to the dimerisation mode of STAT4 via Src-homology (SH2) domains. By concentrating on the residues involved in the intermolecular contacts between STAT5a dimers, it is possible to see that they tend to be highly conserved within STAT5a orthologues, but not between STAT5a and STAT4. Because residues that are located at the interface of protein-protein interactions tend to be conserved, and because in this case most interface residues are of type 3 (specific for the STAT5a and the STAT4 alignment separately), the interaction between the subunits of the STAT5a dimer may be specific.
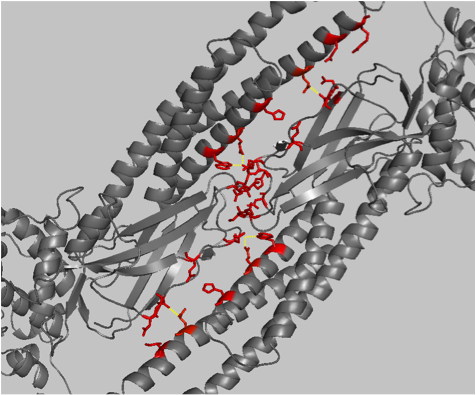
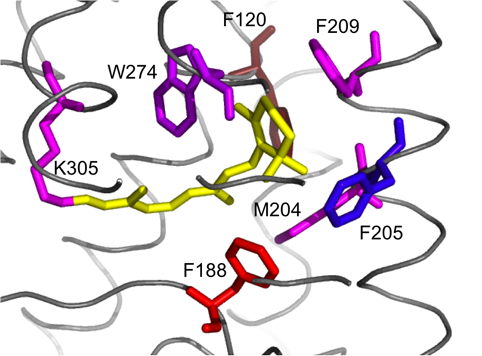
The second example has cephalopod rhodopsins as alignment A and vertebrate rhodopsins as alignment B. Though related, they differ in their molecular properties and function. Whilst vertebrate rhodopsin activates the cyclic GMP signalling pathway, invertebrate rhodopsin activates the inositol-1,4,5-triphosphate signalling pathway via a Gq-type G protein. It is not clear what the cause of the functional difference between these two rhodopsin families is. Our result clearly shows that although some residues that co-ordinate the retinal moiety are conserved between vertebrate and cephalopod rhodopsin, this does not apply to all of them. For example Lys 305, which covalently binds retinal, is conserved between both vertebrate and cephalopod rhodopsin. Other hydrophobic residues in the retinal binding pocket, including Phe 120 and Phe 188, are specific to cephalopod rhodopsin and are not conserved in vertebrate rhodopsin. Phe 205, although it is part of the binding pocket in the squid rhodopsin structure used here, is generally not conserved in cephalopods but in vertebrates.
The sequence logo
The results page will contain a combination of three sequence logos.
The top logo shows positions specific to alignment A, the centre logo shows positions specific to the consensus, and the bottom logo shows positions specific to alignment B.
The sequence alignment
The results page will contain a sequence alignment that combines alignments A and B.
There are two rows that indicate the specificity of each position.
For each position, there are eight possible outcomes, depending on whether the position is specific for the consensus (C), alignment A, alignment B, or any combination of those.
These types are specified in the first row under each alignment.
The types can be summarised into more general categories, which are specified in the second row under each alignment.
Mapping to structure
Optionally, Spial colours the residues of a structure according to whether they are specific to alignment A, alignment B, or both.
Structures can either be taken from PDB or uploaded from your hard disk.
The result is shown on a Jmol web-applet in the results page.
Alternatively, Spial provides scripts that colour structures already opened in PyMOL.
Position types
| type |
category |
specific for... |
explanation |
| C |
A |
B |
| 0 |
0 |
0 |
0 |
0 |
no specificity |
| 1 |
S |
0 |
0 |
1 |
specific for alignment B, but not for alignment A |
| 2 |
S |
0 |
1 |
0 |
specific for alignment A, but not for alignment B |
| 3 |
S |
0 |
1 |
1 |
specific for alignments A and B seperately |
| 4 |
C |
1 |
0 |
0 |
in the consensus only |
| 5 |
C |
1 |
0 |
1 |
in the consensus and specific for alignment B |
| 6 |
C |
1 |
1 |
0 |
in the consensus and specific for alignment A |
| 7 |
C |
1 |
1 |
1 |
in the consensus consensus and specific for alignments A and B |
Each position in an alignment given by Spial can be one of eight possible types.
The type of each position is determined by whether it is specific to alignment A, alignment B, the consensus, or any combination of those.
Each position in an alignment can also be one of three possible categories, which indicate whether the position is specific to the consensus (C), specific to one or both of the input alignments but not the consensus (S), or not specific at all (0).
Types 0, 1, 2, 3 and 4 are self explanatory. Explained below are examples highlighting types 5, 6 and 7 and their biological relevance. Consider a hypothetical protein where the funtionally important residues are marked by '+' sign. Presence of a significant number of type 5 positions would indicate towards the existence of a nested sub-class with sub family B. Similarly, presence of a significant number of type 6 positions would indicate towards the existence of a nested sub-class within the sub-family A. Enrichment for type 7 positions would indicate presence of nested sub-class in the both the sub-families under comparison.
Type 5
# pos 1234567890
+ + + +
seq1.1 AD-RVAT-SH
seq1.2 ADYKV-S-SH
seq1.3 ADY-VVS-SS
seq1.4 ADYGVIS-SS
seq1.5 VDYHVMT-SS
seq2.1 AEWTVMSPSM
seq2.2 ADWTVLSPSM
seq2.3 AEWTMITPPS
seq2.4 ADWTMFTPPS
+ + + +
# type 4531505357
CCSSC0CSCC
|
Type 6
# pos 1234567890
++++
seq1.1 AD-RVATPMS
seq1.2 ADYKV-SPMM
seq1.3 ADY-VVSPSM
seq1.4 ADYGVISMSS
seq1.5 VDYHVMTMSM
seq2.1 AEWTVMSPSM
seq2.2 AEWTVLSPSM
seq2.3 ADWTMISPPS
seq2.4 ADWTMFSPPS
++++
# type 4531506666
CCSSS0CCCC
|
Type 7
# pos 1234567890
++ + ++
seq1.1 AD-RLAT-TH
seq1.2 AQYKV-S-SH
seq1.3 ADY-VVS-SS
seq1.4 VQYGVIS-TS
seq1.5 VDYHLMT-TS
seq2.1 AEWTVMSPSM
seq2.2 ADWTVLSPSM
seq2.3 IDWTMITPPS
seq2.4 IEWTMFTPPS
++ + ++
# type 7731706677
CCSSC0CCCC
|
|
|
|
|
Upload of alignment files
If you upload alignments, files in either SELEX or FASTA format are accepted.
You have to make sure that alignments A and B have the same length (each sequence has the same number of residues).
Empty lines and lines starting with a hash (#) are ignored.
Entering alignments in textboxes
When you input your alignments into the textboxes, make sure they are in SELEX format.
Alignments A and B have to have the same length (each sequence has the same number of residues).
Empty lines and lines starting with a hash (#) are ignored.
Consensus threshold
This option specifies at which proportion an amino acid has to be present in both alignments in order to be considered as consensus.
The number specified has to be between 0 and 1.
See algorithm for details.
Specificity threshold
This option specifies at which proportion a non-consensus amino acid has to be present in an alignment to be considered as specific to this alignment.
The number specified has to be between 0 and 1.
See algorithm for details.
Logo X-axis numbering
With this option you can specify whether you want the logo to have X-axis numbering.
Small sample correction of logo
With this option you can specify whether you want the logo to use small sample correction (rare residues are ignored).
Supplying a PDB ID
If you'd like Spial to get the structure from PDB, enter the 4-letter identifier here.
Uploading PDB files
If you'd like Spial to get upload a structure from your hard disk, enter the file location here.
Query ID in alignment A
This is the name of the sequence in alignment A that corresponds to the sequence of the PDB structure. Spial will return an error message if the two sequences do not match entirely.
Chain
Here you can specify the chain (A, B, C, etc.) of the structure which you'd like to use.
the FASTA format
Example of an alignment in FASTA format:
>Dachshund
QQWTAGIGLLMALIVLLIVAGNVLVIAAI
>Dalmatian
-QWTMGMTMFMAAII--IVMGNIMVIVAI
>Poodle
QQWTAGMGLLVALIV--IVVGNVLVIVAI
the SELEX format
Example of an alignment in SELEX format:
Dachshund QQWTAGIGLLMALIVLLIVAGNVLVIAAI
Dalmatian -QWTMGMTMFMAAII--IVMGNIMVIVAI
Poodle QQWTAGMGLLVALIV--IVVGNVLVIVAI
Jmol
Jmol is a free, open source molecule viewer for students, educators, and researchers in chemistry and biochemistry. It is cross-platform, running on Windows, Mac OS X, and Linux/Unix systems. Spial uses hte Jmol web-applet to display the color coded protein structure. Alternatively, you can use the PyMOL scripts supplied by Spial to visualise the same information on the Pymol standalone software.
PyMOL
To take advantage of the PyMOL scripts provided by Spial, you have to install PyMOL first.
PyMOL is a program to visualise protein structures and can be downloaded here.
Instructions for using the PyMOL scripts supplied by Spial:
Make sure PyMOL is fully installed.
Open your PDB file in PyMOL, then run the scripts either by copying and pasting them into the PyMOL command line or by saving them to your hard drive and then running them via PyMOL's Run command.
This will only work if the sequence of the protein in the PDB file you have loaded into PyMOL is identical with the sequence you have identified as the query ID in the Spial input page.
Comparison with other tools
A comparison of the features of Spial and other similar tools is provided here . The results for the vertebrate-cephalopod rhodopsin family example are provided here for AMAS (click here), SDPpred (click here) and Sequence Harmony (click here).
Related web-servers and tools
References
Chakrabarti S and Panchenko AR. (2009),' Ensemble approach to predict specificity determinants: benchmarking and validation', BMC Bioinformatics, 10:207
Crooks, G. E., et al. (2004), 'WebLogo: a sequence logo generator', Genome Res, 14 (6), 1188-90.
Edgar, R. C. (2004), 'MUSCLE: multiple sequence alignment with high accuracy and high throughput', Nucleic Acids Res, 32 (5), 1792-7.
Horn, F., et al. (2003), 'GPCRDB information system for G protein-coupled receptors', Nucleic Acids Res, 31 (1), 294-7.
Murakami, M. and Kouyama, T. (2008), 'Crystal structure of squid rhodopsin', Nature, 453 (7193), 363-7.
Schneider, T. D. and Stephens, R. M. (1990), 'Sequence logos: a new way to display consensus sequences', Nucleic Acids Res, 18 (20), 6097-100.
Terakita, A. (2005), 'The opsins', Genome Biol, 6 (3), 213.
Vacic, V., Iakoucheva, L. M., and Radivojac, P. (2006), 'Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments', Bioinformatics, 22 (12), 1536-7.